Potassium tert-Butoxide-Catalyzed Dehydrogenative C–H Silylation of Heteroaromatics: A Combined Experimental and Computational Mechanistic Study
Wen-Bo Liu, David P. Schuman, Yun-Fang Yang, Anton A. Toutov, Yong Liang, Hendrik F. T. Klare, Nasri Nesnas, Martin Oestreich, Donna G. Blackmond, Scott C. Virgil, Shibdas Banerjee, Richard N. Zare, Robert H. Grubbs, K. N. Houk, and Brian M. Stoltz
J. Am. Chem. Soc.,
2017, 139, (20), 6867-6879; DOI: 10.1021/jacs.6b13031
06/2017
Building upon the recent Center studies around the potassium tert-butoxide catalyzed C–H silylation of C(sp2)-H bonds, this mechanistic study is the first of two back-to-back articles published this month in JACS. It brings together a collaborative team of 8 faculty level researchers, 4 CCHF members and 4 external to the Center.
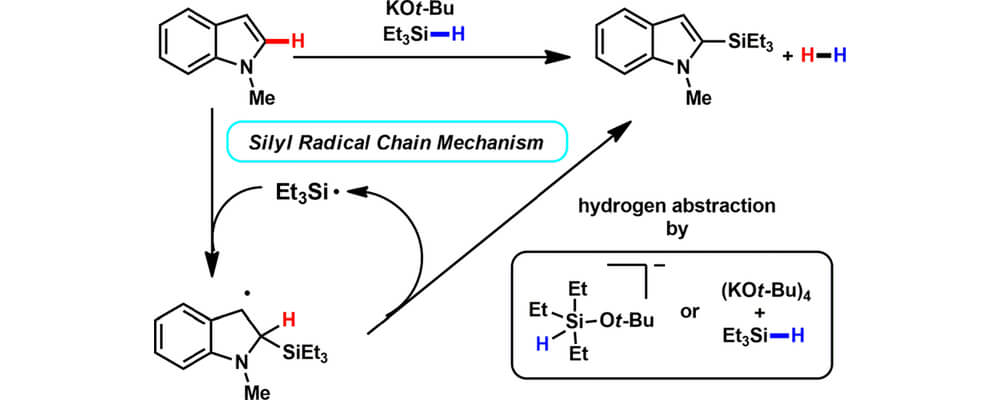
This systematic experimental and computational mechanistic investigation of this transformation, provides results consistent with a radical chain mechanism. A trialkylsilyl radical may be initially generated by homolytic cleavage of a weakened Si–H bond of a hypercoordinated silicon species as detected by IR, or by traces of oxygen which can generate a reactive peroxide by reaction with [KOt-Bu]4 as indicated by density functional theory (DFT) calculations. Radical clock and kinetic isotope experiments support a mechanism in which the C–Si bond is formed through silyl radical addition to the heterocycle followed by subsequent β-hydrogen scission. DFT calculations reveal a reasonable energy profile for a radical mechanism and support the experimentally observed regioselectivity. The silylation reaction is shown to be reversible, with an equilibrium favoring products due to the generation of H2 gas. In situ NMR experiments with deuterated substrates show that H2 is formed by a cross-dehydrogenative mechanism. The stereochemical course at the silicon center was investigated utilizing a 2H-labeled silolane probe; complete scrambling at the silicon center was observed, consistent with a number of possible radical intermediates or hypercoordinate silicates.
Related Content
-
06/2017
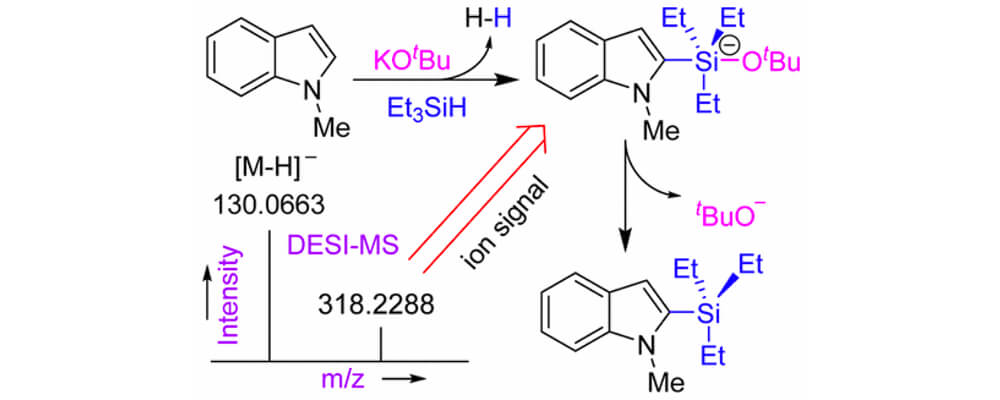
Ionic and Neutral Mechanisms for C–H Bond Silylation of Aromatic Heterocycles Catalyzed by Potassium tert-Butoxide
RESEARCH
-
02/2017
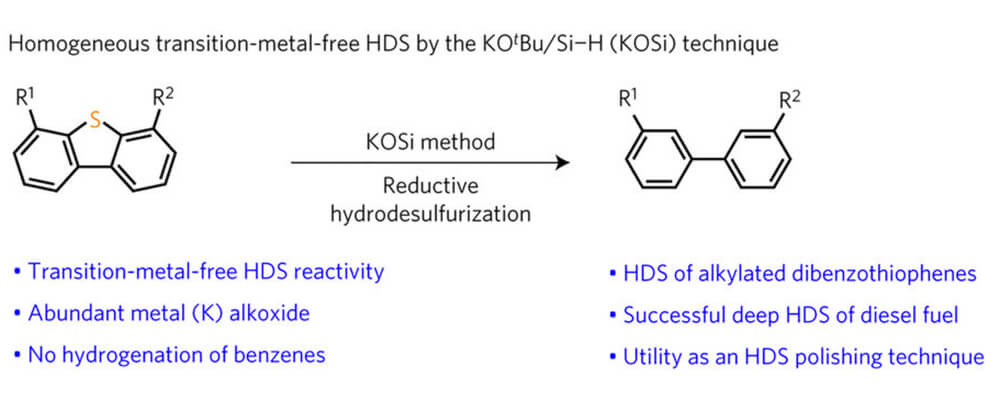
A potassium tert-butoxide and hydrosilane system for ultra-deep desulfurization of fuels
RESEARCH
-
12/2016
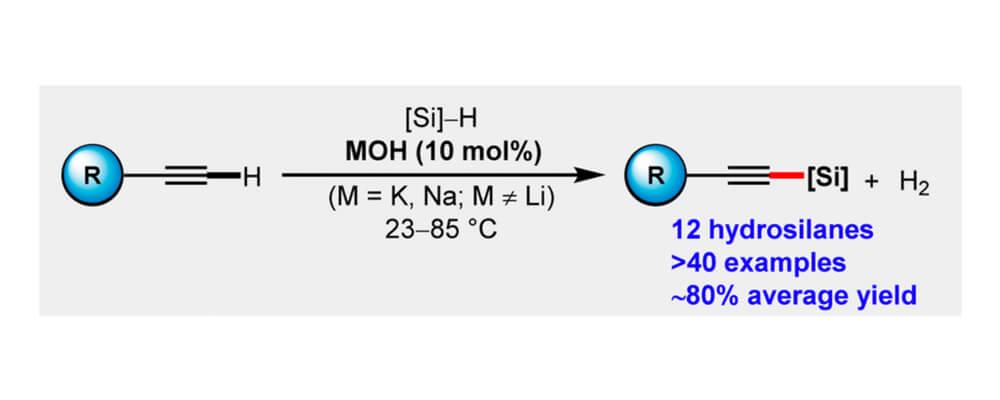
Alkali Metal-Hydroxide-Catalyzed C(sp)–H Bond silylation
RESEARCH
-
11/2016
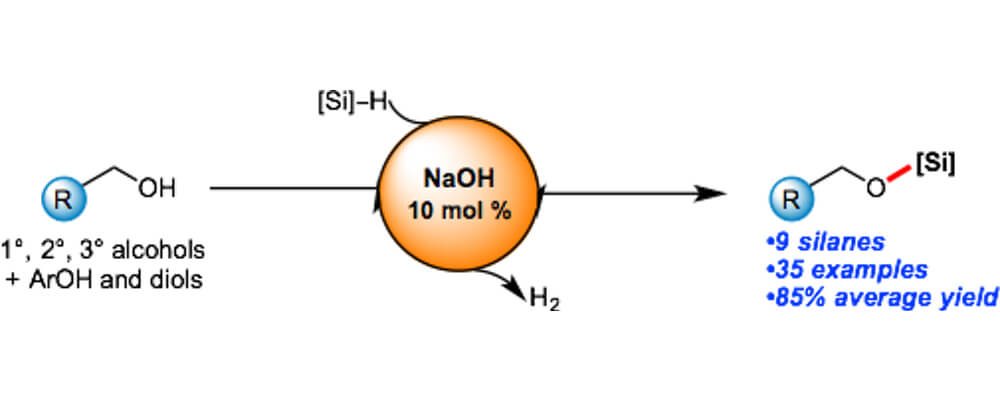
Sodium Hydroxide Catalyzed Dehydrocoupling of Alcohols with Hydrosilanes
RESEARCH
-
08/2016
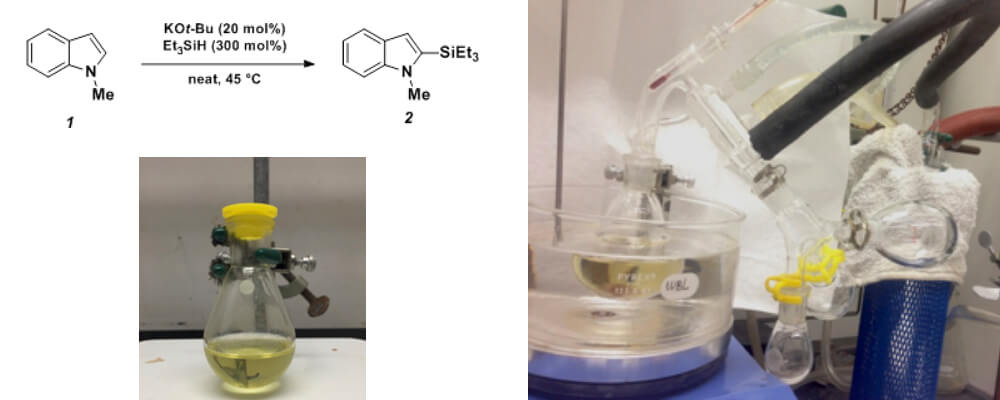
Potassium tert-Butoxide-Catalyzed Dehydrogenative Cross-Coupling of Heteroarenes with Hydrosilanes
RESEARCH
-
10/2015
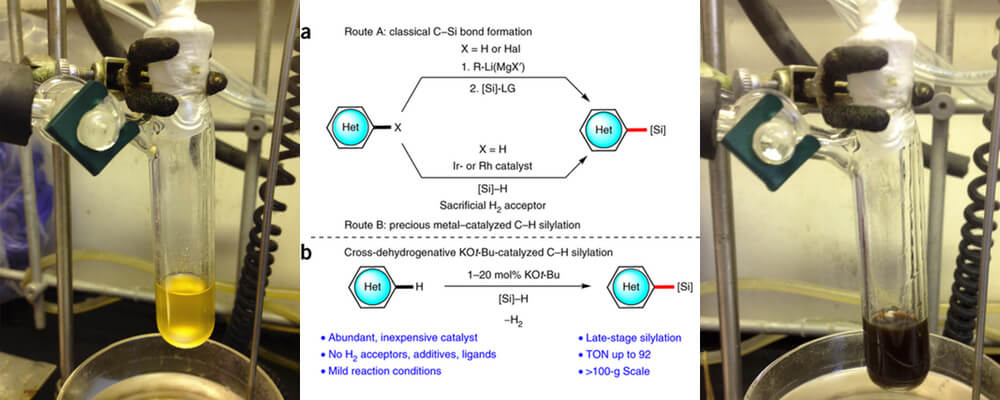
Catalytic C–H bond silylation of aromatic heterocycles
RESEARCH
-

04/2015
Elementary 19 Documentary
OUTREACH
-
01/2015
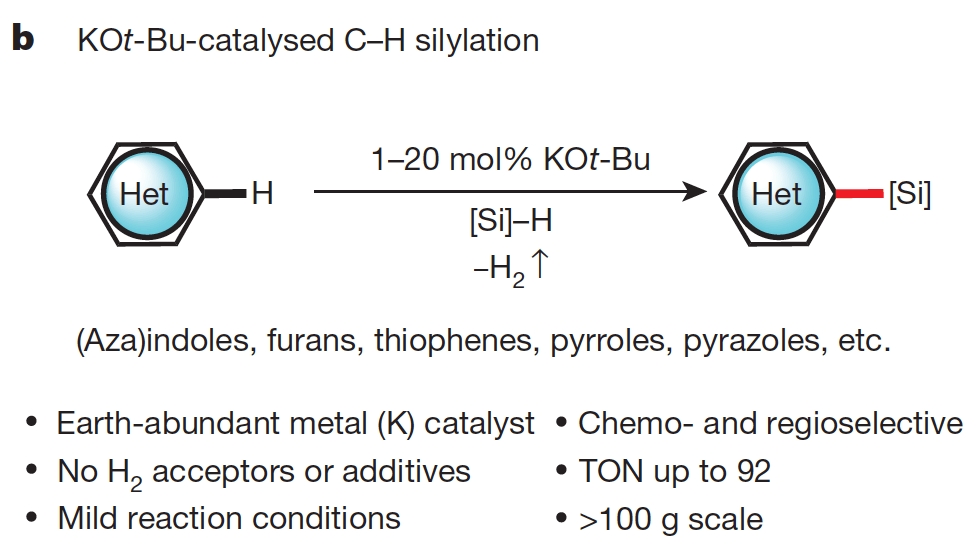
Silylation of C–H bonds in aromatic heterocycles by an Earth-abundant metal catalyst
RESEARCH