Ionic and Neutral Mechanisms for C–H Bond Silylation of Aromatic Heterocycles Catalyzed by Potassium tert-Butoxide
Shibdas Banerjee, Yun-Fang Yang, Ian D. Jenkins, Yong Liang, Anton A. Toutov, Wen-Bo Liu, David P. Schuman, Robert H. Grubbs, Brian M. Stoltz, Elizabeth H. Krenske, Kendall N. Houk, and Richard N. Zare
J. Am. Chem. Soc.,
2017, 139, (20), 6880-6887; DOI: 10.1021/jacs.6b13032
06/2017
Building upon the recent Center studies around the potassium tert-butoxide catalyzed C–H silylation of C(sp2)-H bonds, this mechanistic study is the second of two back-to-back articles published this month in JACS. It brings together a collaborative team of 5 faculty level researchers, 2 CCHF members and 2 external to the Center.
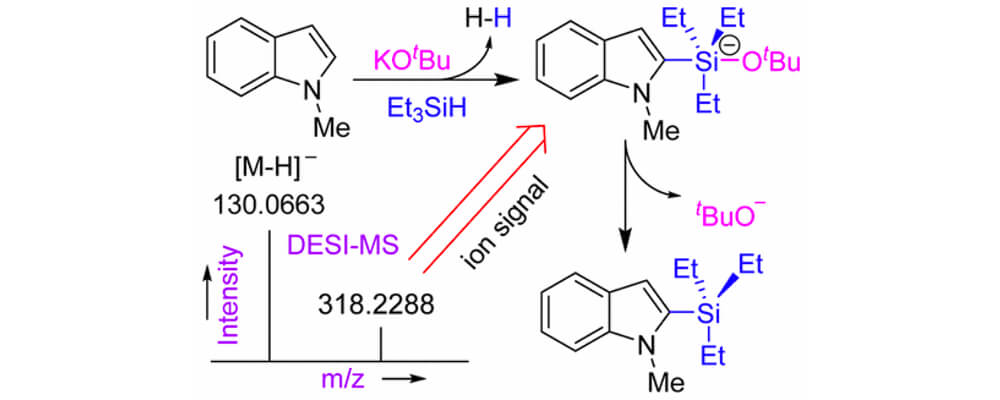
Exploiting C–H bond activation is difficult, although some success has been achieved using precious metal catalysts. Recently, it was reported that C–H bonds in aromatic heterocycles were converted to C–Si bonds by reaction with hydrosilanes under the catalytic action of potassium tert-butoxide alone. The use of Earth-abundant potassium cation as a catalyst for C–H bond functionalization seems to be without precedent, and no mechanism for the process was established. Using ambient ionization mass spectrometry, we are able to identify crucial ionic intermediates present during the C–H silylation reaction. We propose a plausible catalytic cycle, which involves a pentacoordinate silicon intermediate consisting of silane reagent, substrate, and the tert-butoxide catalyst. Heterolysis of the Si–H bond, deprotonation of the heteroarene, addition of the heteroarene carbanion to the silyl ether, and dissociation of tert-butoxide from silicon lead to the silylated heteroarene product. The steps of the silylation mechanism may follow either an ionic route involving K+ and tBuO– ions or a neutral heterolytic route involving the [KOtBu]4 tetramer. Both mechanisms are consistent with the ionic intermediates detected experimentally. We also present reasons why KOtBu is an active catalyst whereas sodium tert-butoxide and lithium tert-butoxide are not, and we explain the relative reactivities of different (hetero)arenes in the silylation reaction. The unique role of KOtBu is traced, in part, to the stabilization of crucial intermediates through cation−π interactions.
Related Content
-
06/2017
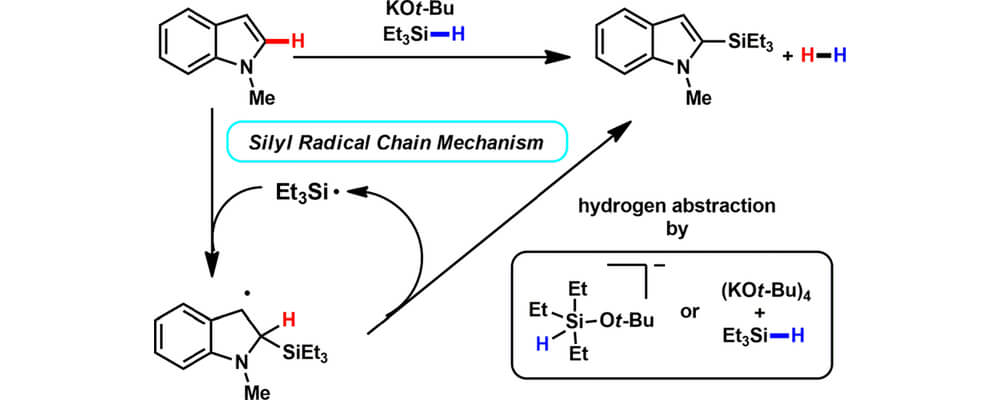
Potassium tert-Butoxide-Catalyzed Dehydrogenative C–H Silylation of Heteroaromatics: A Combined Experimental and Computational Mechanistic Study
RESEARCH
-
02/2017
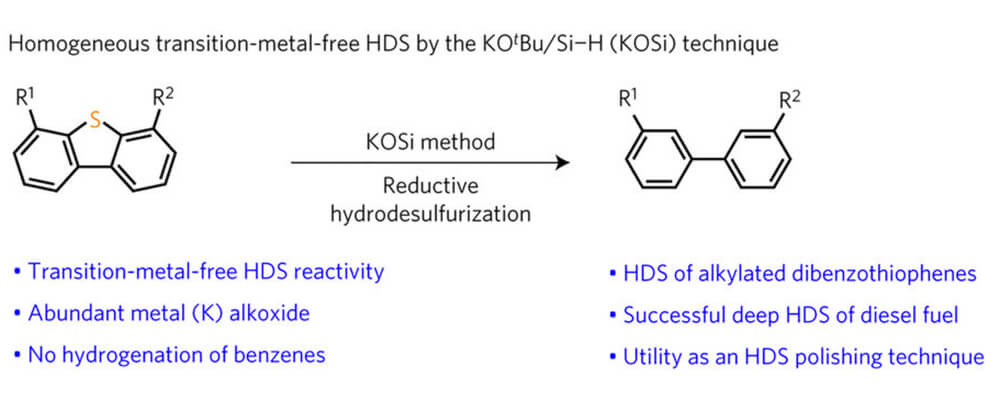
A potassium tert-butoxide and hydrosilane system for ultra-deep desulfurization of fuels
RESEARCH
-
12/2016
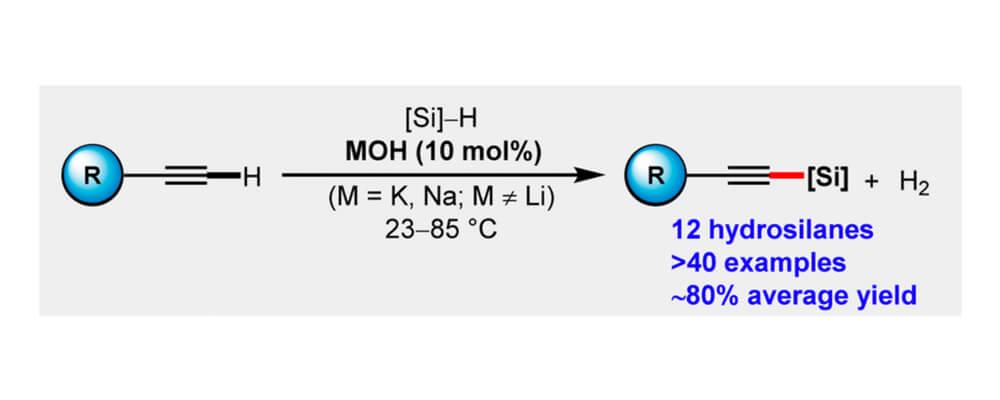
Alkali Metal-Hydroxide-Catalyzed C(sp)–H Bond silylation
RESEARCH
-
11/2016
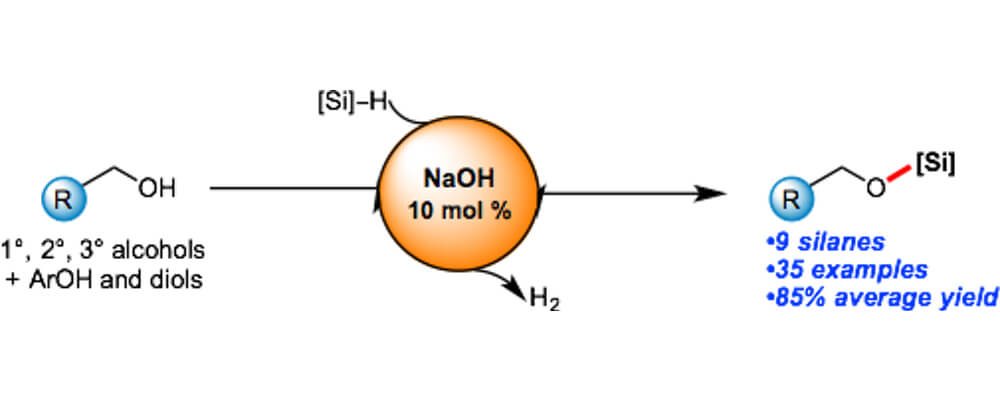
Sodium Hydroxide Catalyzed Dehydrocoupling of Alcohols with Hydrosilanes
RESEARCH
-
08/2016
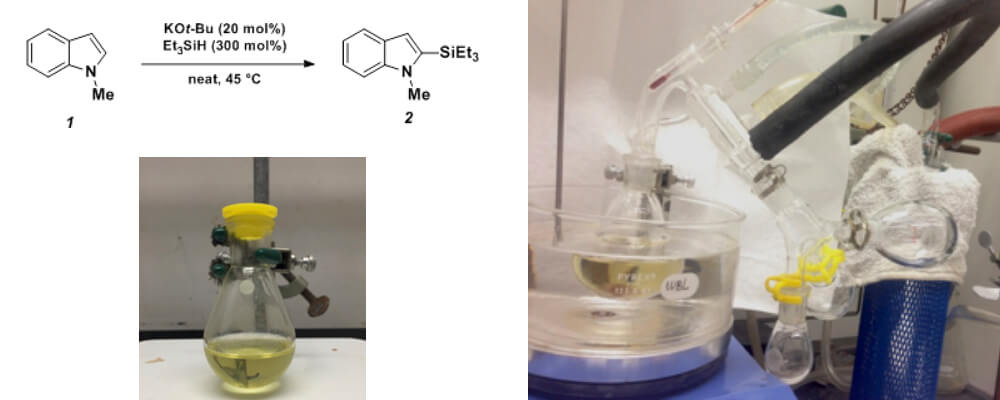
Potassium tert-Butoxide-Catalyzed Dehydrogenative Cross-Coupling of Heteroarenes with Hydrosilanes
RESEARCH
-
10/2015
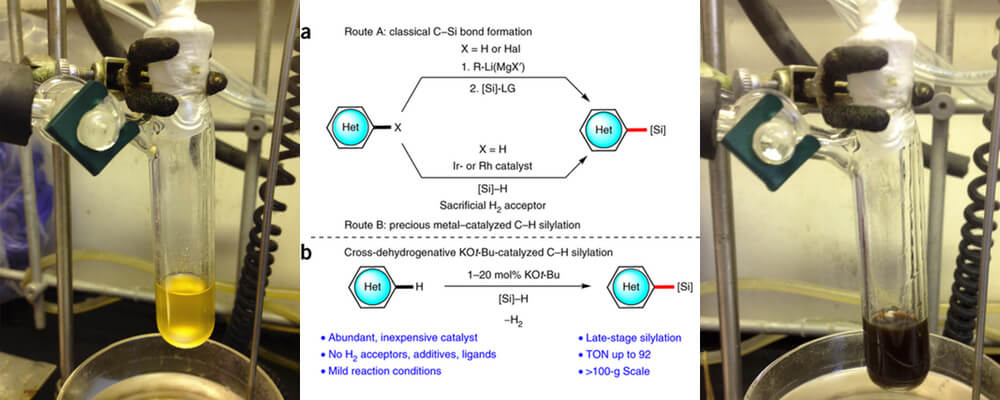
Catalytic C–H bond silylation of aromatic heterocycles
RESEARCH
-

04/2015
Elementary 19 Documentary
OUTREACH
-
01/2015
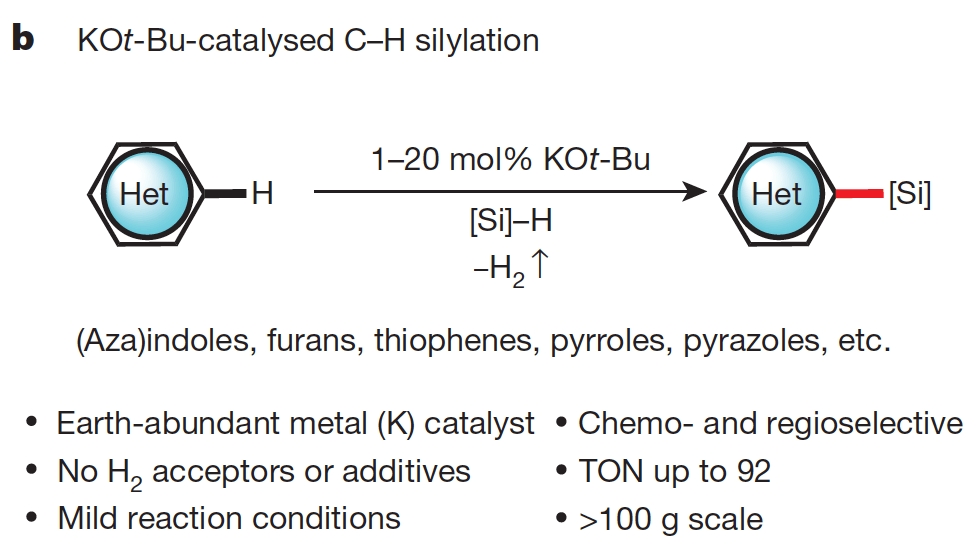
Silylation of C–H bonds in aromatic heterocycles by an Earth-abundant metal catalyst
RESEARCH