Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics through Late-Stage Polyketide Assembly, Tailoring, and C—H Functionalization
Andrew N. Lowell, Matthew D. DeMarsII, Samuel T. Slocum, Fengan Yu, Krithika Anand, Joseph A. Chemler, Nisha Korakavi, Jennifer K. Priessnitz, Sung Ryeol Park, Aaron A. Koch, Pamela J. Schultz, and David H. Sherman
J. Am. Chem. Soc.,
2017, 139, (23), 7913-7920; DOI:10.1021/jacs.7b02875
05/2017
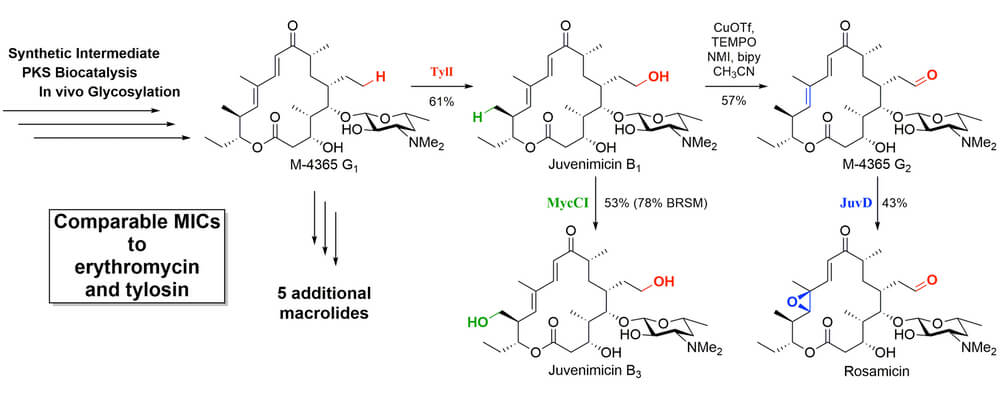
Functionalization of an unactivated C-H bond in the presence of other bonds with similar reactivity represents the ultimate feat of selectivity. Chemoenzymatic methods, where a biological system has been designed as a catalyst capable of selecting a single center in a highly functionalized, unprotected system, offer a way to this goal.
This article from the Sherman group uses synthetic chemistry and biocatalysis to chemoenzymatically achieve the first total synthesis of the juvenimicin/M-4365/rosamicin family of macrolide antibiotics. By leveraging the unique reactivity of cytochrome P450 monooxygenases, a single macrolide intermediate was diversified into the entire family of differentially oxidized natural products. TylI (red) created hydroxyl functionality on an unactivated ethyl chain that could be further oxidized under standard chemical conditions; MycCI (green) hydroxylated an unactivated methyl group; JuvD (blue) selectively epoxidized the – double bond. These transformations relied on the directing power of the P450 catalyst, thus avoiding the need to covalently attach a directing group to the target substrate or rely on its inherent reactivity profile. Testing of the flexibility of these remarkable catalysts on other systems is underway.