Rhodium(II)-Catalyzed C–H Functionalization of Electron-Deficient Methyl Groups
Liangbing Fu, David M. Guptill, and Huw M. L. Davies
J. Am. Chem. Soc.,
2016, 138 (18), 5761-5764; 10.1021/jacs.6b01941
04/2016
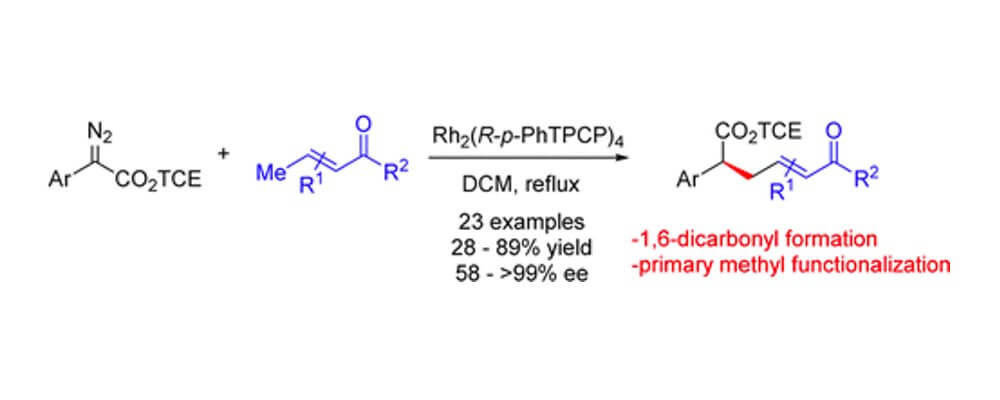
Intermolecular C–H insertion of donor/acceptor carbenes has been established as a highly efficient and selective transformation. Through extensive mechanistic studies it has been demonstrated that the highly electrophilic character of the carbene leads the reaction to proceed through an concerted asynchronous transition state, characterized by a buildup of positive charge at the carbon of the C–H bond being functionalized. Hence electron-rich C–H bonds have been the most preferred sites for selection, such as allylic, benzyllic or those adjacent to heteroatoms.
This study demonstrates the control possible with catalyst design and judicious reagent choice. Using the more robust TCE-ester diazo compounds and the TPCP class of dirhodium catalysts primary, electron-deificient C–H bonds on crotyl substrate is possible. The sterically hindered nature of these new catalysts enables effective reaction at a position that would simply not be possible using the more established conditions.
This transformation adds to the toolbox of carbene C–H insertion reactions, furnishing an efficient and highly stereoselective entry into 1,6-dicarbonyl structures.