Mono-N-protected amino acid ligands stabilize dimeric palladium(II) complexes of importance to C–H functionalization
Joseph J. Gair, Brandon E. Haines, Alexander S. Filatov, Djamaladdin G. Musaev and Jared C. Lewis
Chemical Science
2017, 8, 5746-5756; DOI:10.1039/C7SC01674C
06/2017
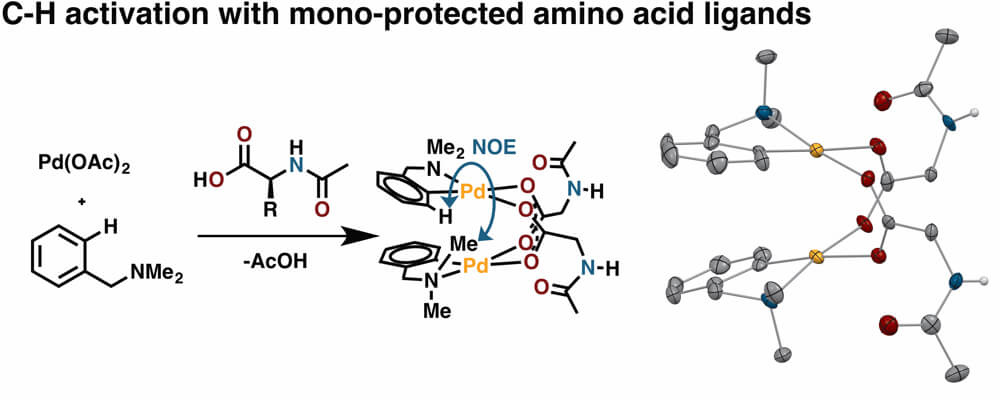
The discovery within the Center for Selective C-H Functionalization (CCHF) that mono-protected amino acid (MPAA) ligands can dramatically improve rates, yields, and selectivities of palladium catalyzed C-H functionalization reactions has motivated a number of collaborative studies to illuminate the role of MPAAs in these remarkable transformations. These combined efforts have made great strides toward establishing how MPAA ligands influence rates and selectivities in palladium catalyzed C-H functionalization reactions; however, studies of the structure and reactivity of isolated Pd/MPAA complexes remain sparse.
This collaboration between the Lewis and Musaev groups adds to the collective effort in the CCHF to understand and leverage the influence of MPAA ligands in C-H activation by exploring the structure and reactivity of isolated Pd/MPAA complexes. Studying isolated complexes—instead of catalytic outcomes— provides a new perspective which both corroborates and offers exciting alternatives to the current model for how MPAA ligands enable remarkable C-H functionalizations.
Our experimental and computational studies of C-H activation reactions with MPAA ligands confirm that MPAAs are strong ligands capable of displacing acetate— in line with the current model derived from catalytic outcomes. Exchange experiments are consistent with the presence of monomeric MPAA complexes. In contrast to the current model, our experimental and computational studies suggest that these complexes exist as carboxylate bridged dimers with high energy monomeric species present in rapid equilibrium— for this particular combination of reaction conditions, MPAA, and substrate. Surprisingly, the chirality of stacked, square planar palladium dimers results in a mixture of diastereomeric complexes in the presence of chiral MPAAs. This unexpected diastereotopicity (with a substrate lacking stereogenic centers) reveals that bridging MPAA ligands convey stereochemical information, which is maintained via stronger binding of bridged MPAAs relative to acetate.