Analyzing Site Selectivity in Rh2(esp)2-Catalyzed Intermolecular C–H Amination Reactions
Elizabeth N. Bess, Ryan J. DeLuca, Daniel J. Tindall, Martins S. Oderinde, Jennifer L. Roizen, J. Du Bois, and Matthew S. Sigman
J. Am. Chem. Soc.,
2014, 136, (15), 5783-5789; 10.1021/ja5015508
03/2014
Conventional synthetic organic chemistry relies upon large differences in bond dissociation energies in order to regulate selective reactions, however, with the advent of C–H Functionalization the difference between different reactive sites (i.e. different C–H bonds) becomes significantly smaller, which in turn means that the factors that impact selectivity in these reactions become more subtle.
A recent publication in the Journal of the American Chemical Society highlights a new technique used to try and untangle these subtle factors.
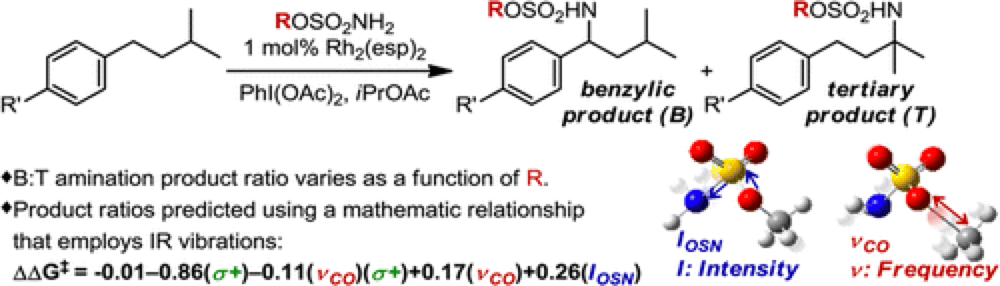
The Du Bois group have recently reported an intermolecular rhodium(II)-catalyzed C–H Amination protocol that demonstrates a site-selectivity between a benzylic and tertiary position that is highly dependent upon the choice of sulfamate ester reagent employed. The relationships between the steric and electronic factors of sulfamate ester and the reaction outcome are not obvious.
The classic physical organic chemistry technique used to delineate the properties that influence reaction outcome is linear free-energy relationship (LFER) analysis. This technique was pioneered by Hammett for electronic analysis of aromatic substrates and further developed by Taft and Charton for steric effect analysis. These techniques have significant limitations in terms of reaction scope and the level of detail they impart.
The Sigman group have very recently reported a new technique, based upon the discovery that specific infrared (IR) molecular vibrations represent an applicable and descriptive parameter set that can predictably calculated for any molecule.
Starting with a training set of sulfamate esters, identifying a group of IR vibration parameters and using stepwise linear regression analysis a subset of parameters that best mathematically relate the features of the sulfamate ester with the energy difference in site-selection were derived. These were used to develop a mathematical model of this reaction which was found to correlate closely to the experimental results.
This not only describes the factors responsible for site selectivity in this transformation but can also be used to predict reaction selectivity, showcased by the development of three new sulfamate esters that furnish excellent site selectivity.
This modeling approach will be applied to examine other selectivity challenges within the CCHF.